
用scanpy对pbmc示例数据进行单细胞数据分析流程学习
下载数据集
在终端中依次输入下列命令下载PBMC3K演示数据集。
mkdir data
wget http://cf.10xgenomics.com/samples/cell-exp/1.1.0/pbmc3k/pbmc3k_filtered_gene_bc_matrices.tar.gz -O data/pbmc3k_filtered_gene_bc_matrices.tar.gz
cd data; tar -xzf pbmc3k_filtered_gene_bc_matrices.tar.gz
mkdir write
加载必要的库
import scanpy as sc
import pandas as pd
import anndata as ad
import numpy as np
import matplotlib.pyplot as plt
设置图形的参数,包括设置DPI为80(图像分辨率)和背景颜色为白色。这些参数将应用于后续生成的图形。
sc.settings.verbosity = 3 # 详细程度:错误 (0), 警告 (1), 信息 (2), 提示 (3)
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor="white")
scanpy==1.10.1 anndata==0.10.7 umap==0.5.5 numpy==1.26.4 scipy==1.13.1 pandas==2.2.2 scikit-learn==1.5.0 statsmodels==0.14.2 igraph==0.11.8 pynndescent==0.5.13
读取数据
results_file = "write/pbmc3k.h5ad" # 将存储分析结果的文件
adata = sc.read_10x_mtx(
"data/filtered_gene_bc_matrices/hg19/", # 包含`.mtx`文件的目录
var_names="gene_symbols", # 使用基因符号作为变量名(变量轴索引)
cache=True, # 写入缓存文件以加快后续读取速度
)
adata #查看数据
adata.var_names_make_unique() # 确保数据中的变量名(基因名)是唯一的
... reading from cache file cache/data-filtered_gene_bc_matrices-hg19-matrix.h5ad
可以看到原始文件genes.tsv中有两列,分别是symbol_id和ENSEMBL_ID,vars_names = “gene_symbols”则可以直接将基因的索引设置为symbol_id,这一点似乎比seurat对象的操作更为方便。
读取tsv文件时可以继续用pd.read_csv()函数,并指定sep=’\t’参数,因为tsv文件是用制表符分隔的,而不是逗号。
temp_vars = pd.read_csv('data/filtered_gene_bc_matrices/hg19/genes.tsv',sep = '\t',header = None)
temp_vars.head()
| 0 | 1 | |
|---|---|---|
| 0 | ENSG00000243485 | MIR1302-10 |
| 1 | ENSG00000237613 | FAM138A |
| 2 | ENSG00000186092 | OR4F5 |
| 3 | ENSG00000238009 | RP11-34P13.7 |
| 4 | ENSG00000239945 | RP11-34P13.8 |
质量控制(QC)
在 scanpy 库中,pl 和 pp 是两个常用的子模块,分别用于绘图(plotting)和预处理(preprocessing)。
pl 模块(Plotting)
示例函数: sc.pl.highest_expr_genes(adata, n_top=20): 绘制表达量最高的20个基因的柱状图。
sc.pl.scatter(adata, x=’n_counts’, y=’percent_mito’): 绘制细胞总基因数与线粒体基因比例的散点图。
sc.pl.heatmap(adata, var_names=gene_list, groupby=’cluster’): 绘制基因表达的热图。
sc.pl.umap(adata, color=[‘cluster’, ‘gene_name’]): 绘制UMAP降维后的细胞分布图,并着色显示不同的聚类结果和特定基因的表达情况。
pp 模块(Preprocessing)
pp 模块提供了各种预处理函数,用于清理和准备单细胞RNA测序数据,以便进行进一步的分析。这些函数包括数据过滤、归一化、特征选择等。
示例函数: sc.pp.filter_cells(adata, min_genes=200): 过滤掉表达基因数少于200的细胞。
sc.pp.filter_genes(adata, min_cells=3): 过滤掉在少于3个细胞中表达的基因。
sc.pp.normalize_total(adata, target_sum=1e4): 对数据进行全局归一化,使得每个细胞的总表达量为1e4。
sc.pp.log1p(adata): 对数据进行log1p变换,以减少数据的偏斜。
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5): 选择高变基因,这些基因在不同细胞间表现出较大的表达变异,是后续分析的重要特征。
sc.pp.scale(adata, max_value=10): 对数据进行缩放处理,使其具有相似的方差。
# 预处理
sc.pl.highest_expr_genes(adata, n_top=20) # 查看表达量最高的20个基因
sc.pp.filter_cells(adata, min_genes=200) # 过滤掉小于200个基因的细胞
sc.pp.filter_genes(adata, min_cells=3) # 过滤掉小于3个细胞的基因
# 计算线粒体基因比例
adata.var["mt"] = adata.var_names.str.startswith("MT-") # 将线粒体基因组标记为'mt'
sc.pp.calculate_qc_metrics(
adata, qc_vars=["mt"], percent_top=None, log1p=False, inplace=True
)
normalizing counts per cell
finished (0:00:02)
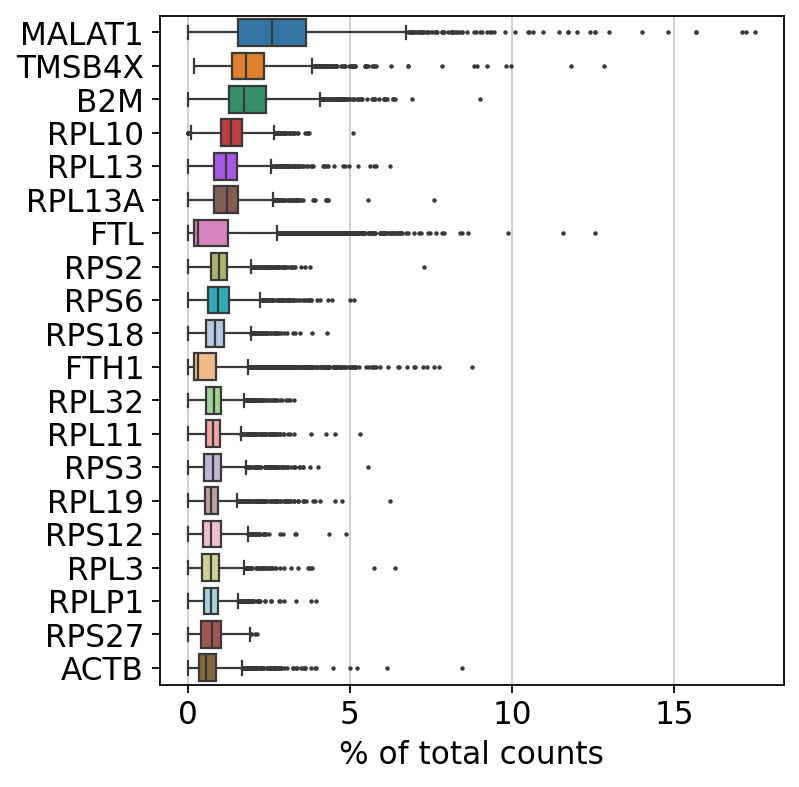
filtered out 19024 genes that are detected in less than 3 cells
adata.obs.columns
Index(['n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt',
'pct_counts_mt'],
dtype='object')
可以看到目前adata中有5个obs了,即对应的每一个细胞的元数据,展示每一个细胞在这几个维度上的表现。
sc.pl.violin(
adata,
["n_genes_by_counts", "total_counts", "pct_counts_mt",],
jitter=0.4,
multi_panel=True,
)
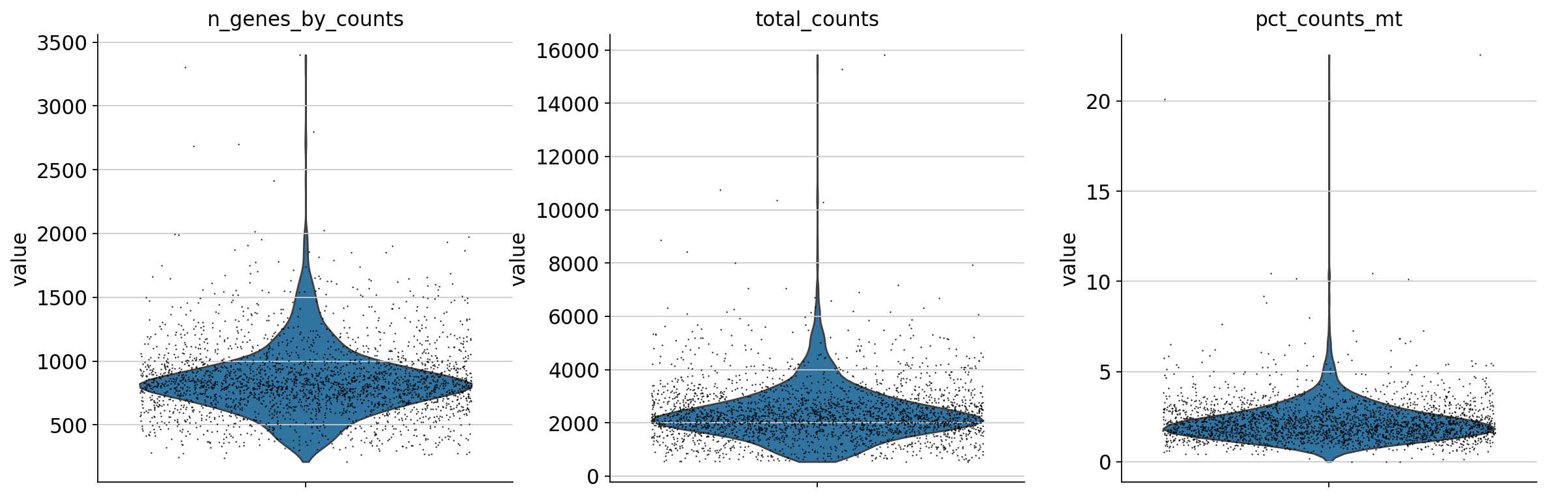
注意下面的图形用matplotlib进行子图的排列,以达到类似R中patchwork的效果。
#去除线粒体基因表达过多或总数过多的细胞:
fig, axs = plt.subplots(1,2, figsize=(10,5))
sc.pl.scatter(adata, x="total_counts", y="pct_counts_mt", ax=axs[0],show = False) # 总计数与线粒体基因比例散点图
axs[0].set_title('test1')
sc.pl.scatter(adata, x="total_counts", y="n_genes_by_counts",ax=axs[1],show = False) # 总计数与基因表达量散点图
axs[1].set_title('test2')
plt.tight_layout()
plt.show()
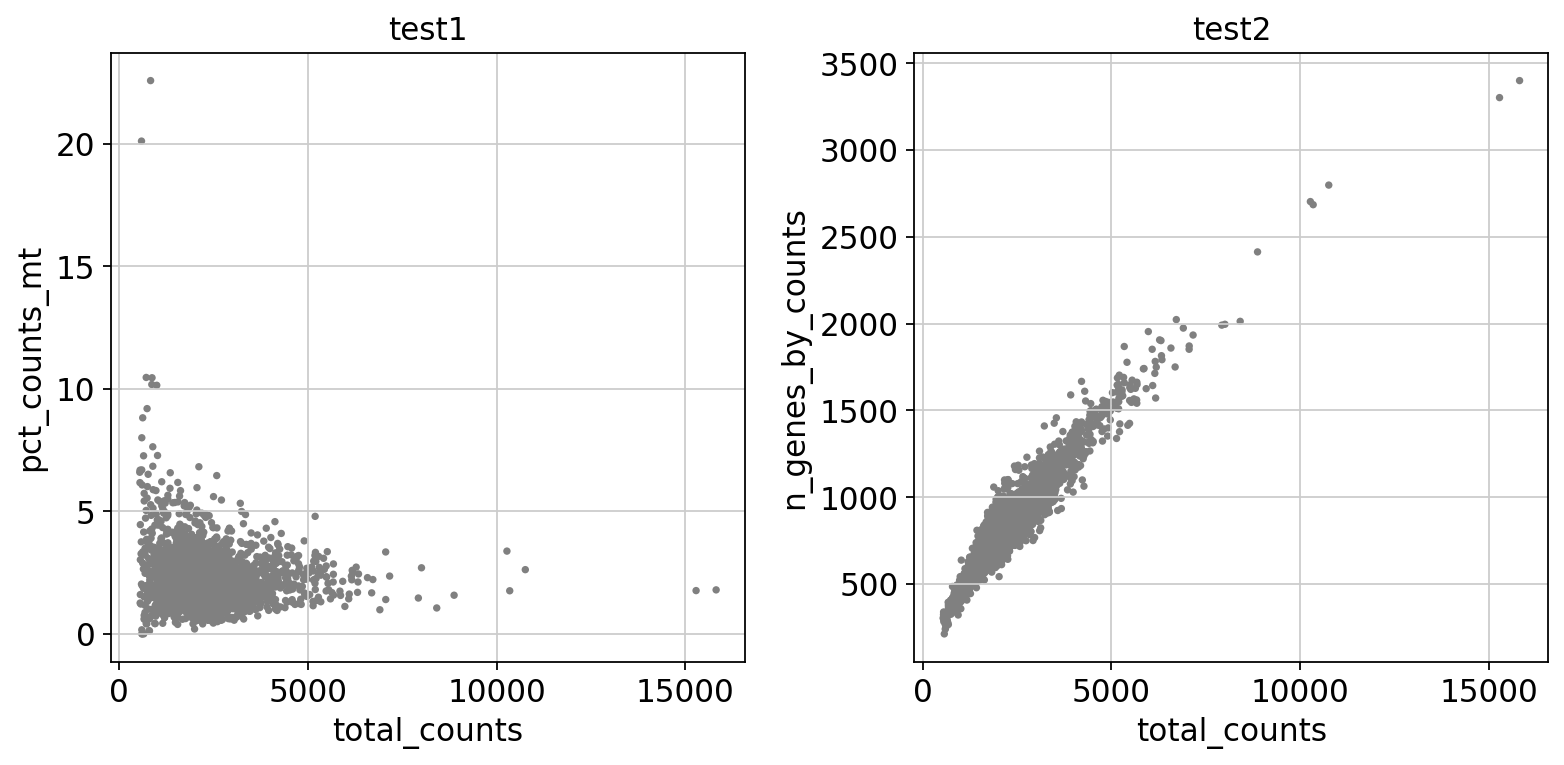
#实际上通过切片AnnData对象来进行过滤
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :].copy()
sc.pp.normalize_total(adata, target_sum=1e4) # 标准化细胞总计数
sc.pp.log1p(adata) # 对数变换
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5) #识别高变异基因
sc.pl.highly_variable_genes(adata) # 绘制高变异基因的分布图
normalizing counts per cell
finished (0:00:01)
extracting highly variable genes
finished (0:00:03)
--> added
'highly_variable', boolean vector (adata.var)
'means', float vector (adata.var)
'dispersions', float vector (adata.var)
'dispersions_norm', float vector (adata.var)
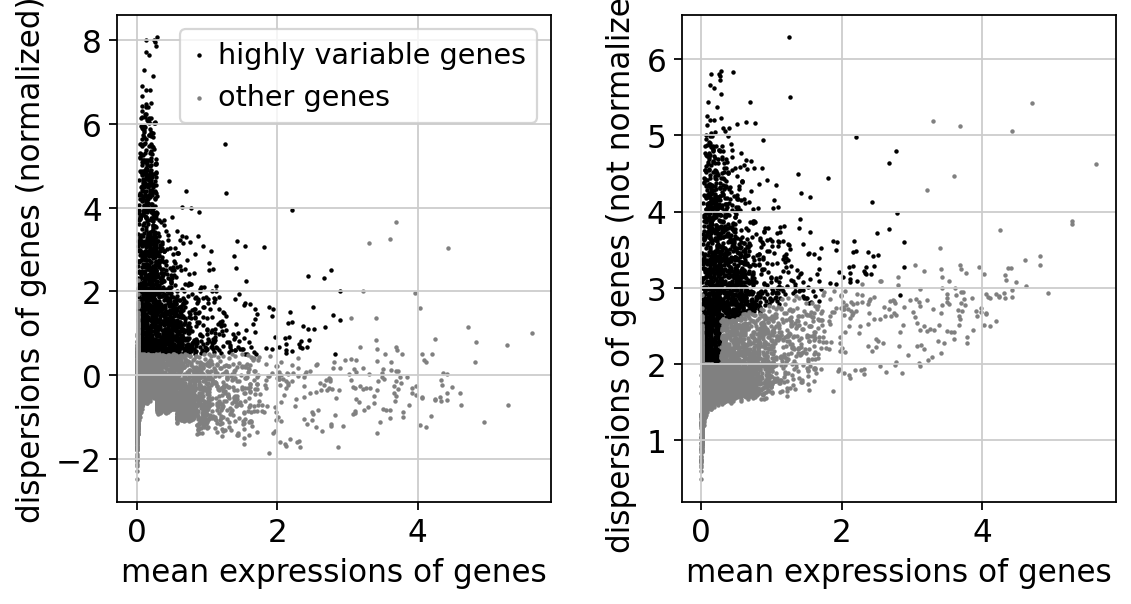
将AnnData对象的属性设置为标准化和对数化的原始基因表达,以便以后在基因表达的差异测试和可视化中使用。这只是冻结AnnData对象的状态。
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
sc.pp.regress_out(adata, ["total_counts", "pct_counts_mt"]) # 回归除线粒体基因和总计数之外的其他因素
sc.pp.scale(adata, max_value=10) # 标准化基因表达量
regressing out ['total_counts', 'pct_counts_mt']
sparse input is densified and may lead to high memory use
/home/woodpecker/anaconda3/envs/py3.10/lib/python3.10/site-packages/scanpy/preprocessing/_simple.py:641: UserWarning: Received a view of an AnnData. Making a copy.
view_to_actual(adata)
finished (0:00:25)
降维聚类和差异分析
# 主成分分析
sc.tl.pca(adata, svd_solver="arpack") # 主成分分析
computing PCA
with n_comps=50
finished (0:00:55)
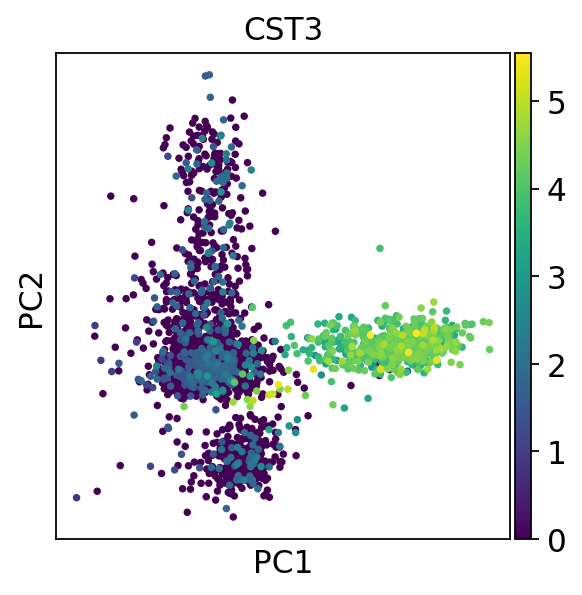
sc.pl.pca(adata, color="CST3") # 绘制主成分分析结果
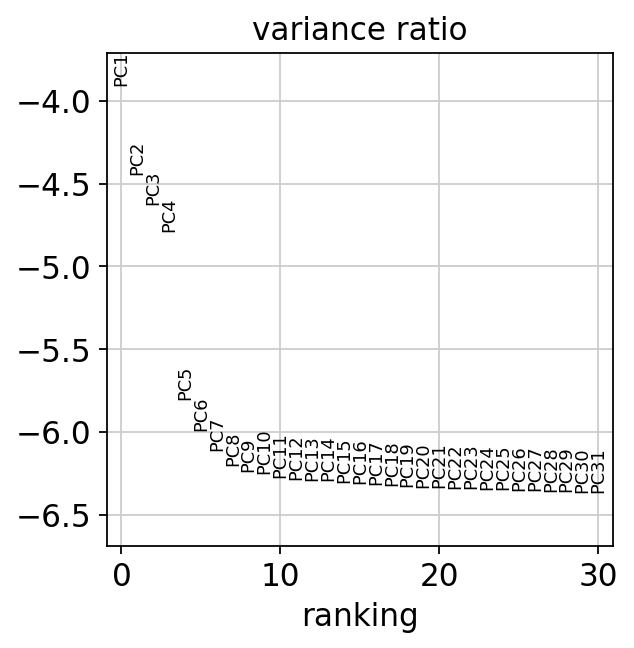
sc.pl.pca_variance_ratio(adata, log=True)
sc.tl.leiden(
adata,
resolution=0.9,
random_state=0,
flavor="igraph",
n_iterations=2,
directed=False,
)
running Leiden clustering
finished: found 8 clusters and added
'leiden', the cluster labels (adata.obs, categorical) (0:00:00)
这里的leiden方法类似于Seurat中的FindClusters方法,但速度更快,根据前面计算的距离矩阵,使用聚类算法对数据进行聚类,找到合适的cluster数目,在这里也可以设定分辨率参数resolution,其值越高,聚类后的cluster越多。
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
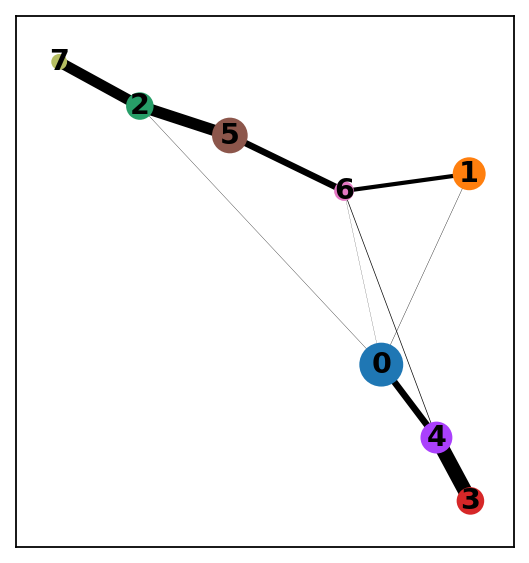
sc.tl.paga(adata)
sc.pl.paga(adata) # remove `plot=False` if you want to see the coarse-grained graph
sc.tl.umap(adata, init_pos='paga')
sc.tl.umap(adata)
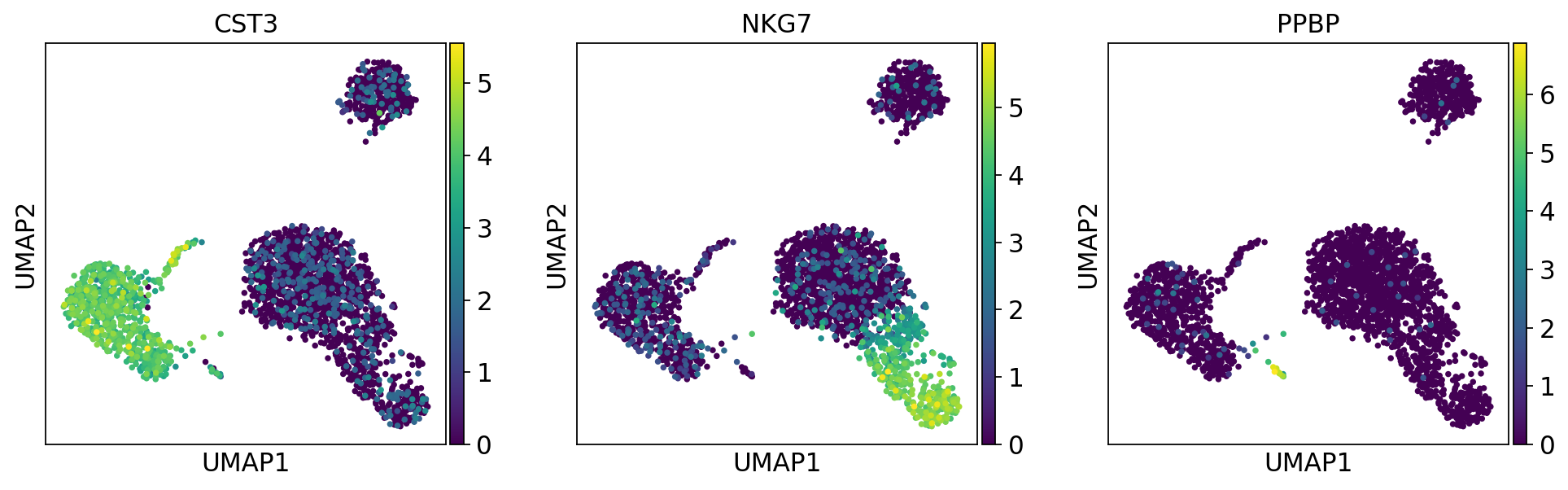
sc.pl.umap(adata, color=["CST3", "NKG7", "PPBP"])
computing neighbors
using 'X_pca' with n_pcs = 40
finished: added to `.uns['neighbors']`
`.obsp['distances']`, distances for each pair of neighbors
`.obsp['connectivities']`, weighted adjacency matrix (0:00:01)
running PAGA
finished: added
'paga/connectivities', connectivities adjacency (adata.uns)
'paga/connectivities_tree', connectivities subtree (adata.uns) (0:00:00)
--> added 'pos', the PAGA positions (adata.uns['paga'])
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:08)
computing UMAP
finished: added
'X_umap', UMAP coordinates (adata.obsm) (0:00:09)
# sc.tl.tsne(adata)
sc.pl.tsne(adata, color = 'leiden')
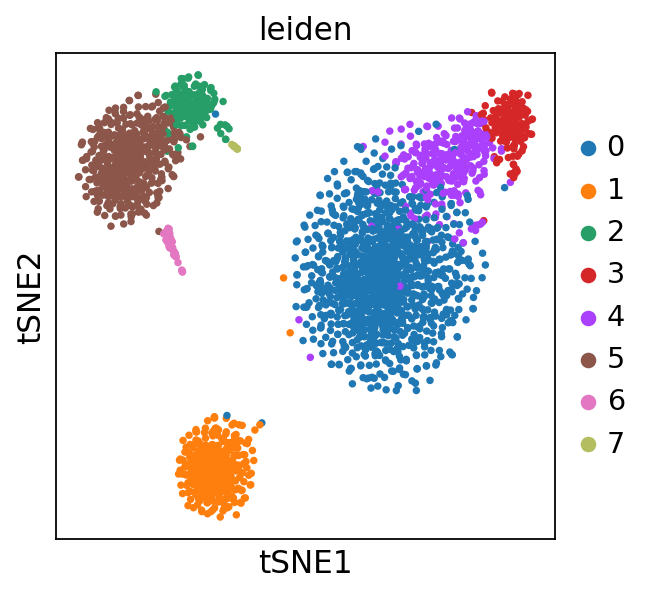
在leiden聚类的基础上进行umap或者tsne降维可视化,在obsm中会生成降维后的坐标信息,可直接使用,进行可视化,这与Seurat中的RunUMAP()和RunTSNE()函数类似。
寻找各个Cluster中的特征性表达基因GeneMarkers
用t检验的方法,对每个Cluster中的基因表达量进行统计检验,找出显著性显著的基因作为特征性表达基因。
sc.tl.rank_genes_groups(adata, "leiden", method="t-test")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:01)
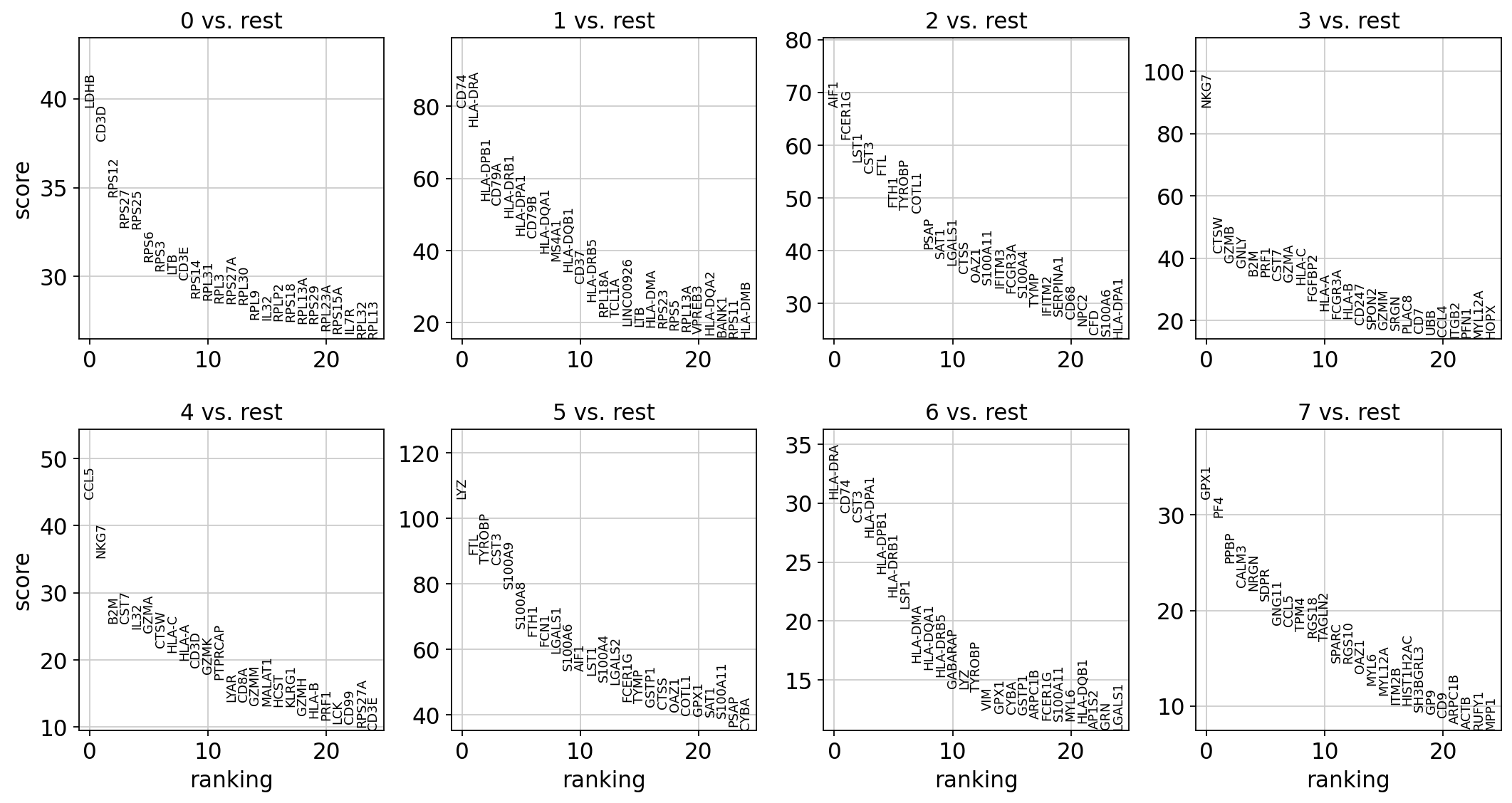
这段代码的主要功能是对单细胞数据进行差异基因表达分析,并可视化不同细胞群之间的差异表达基因。通过Leiden聚类算法确定细胞群后,使用t-test方法计算每个细胞群与其它细胞群之间差异表达的基因,并最终在每个细胞群中展示表达差异最显著的25个基因。
用秩和检验wilcoxon也能得到类似的结果,scanpy官方建议使用后者。
sc.settings.verbosity = 2 # reduce the verbosity
这段代码的主要功能是配置Scanpy库的详细输出级别,以便在数据分析过程中控制输出信息的详细程度,从而帮助用户更好地监控程序的运行状态。具体来说,sc.settings.verbosity = 2将详细程度级别设置为2,这意味着在执行某些操作时,程序会输出相对较少的信息,但仍然会提供一些有用的反馈,以帮助用户了解程序的运行状态。
sc.tl.rank_genes_groups(adata, "leiden", method="wilcoxon")
sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:06)
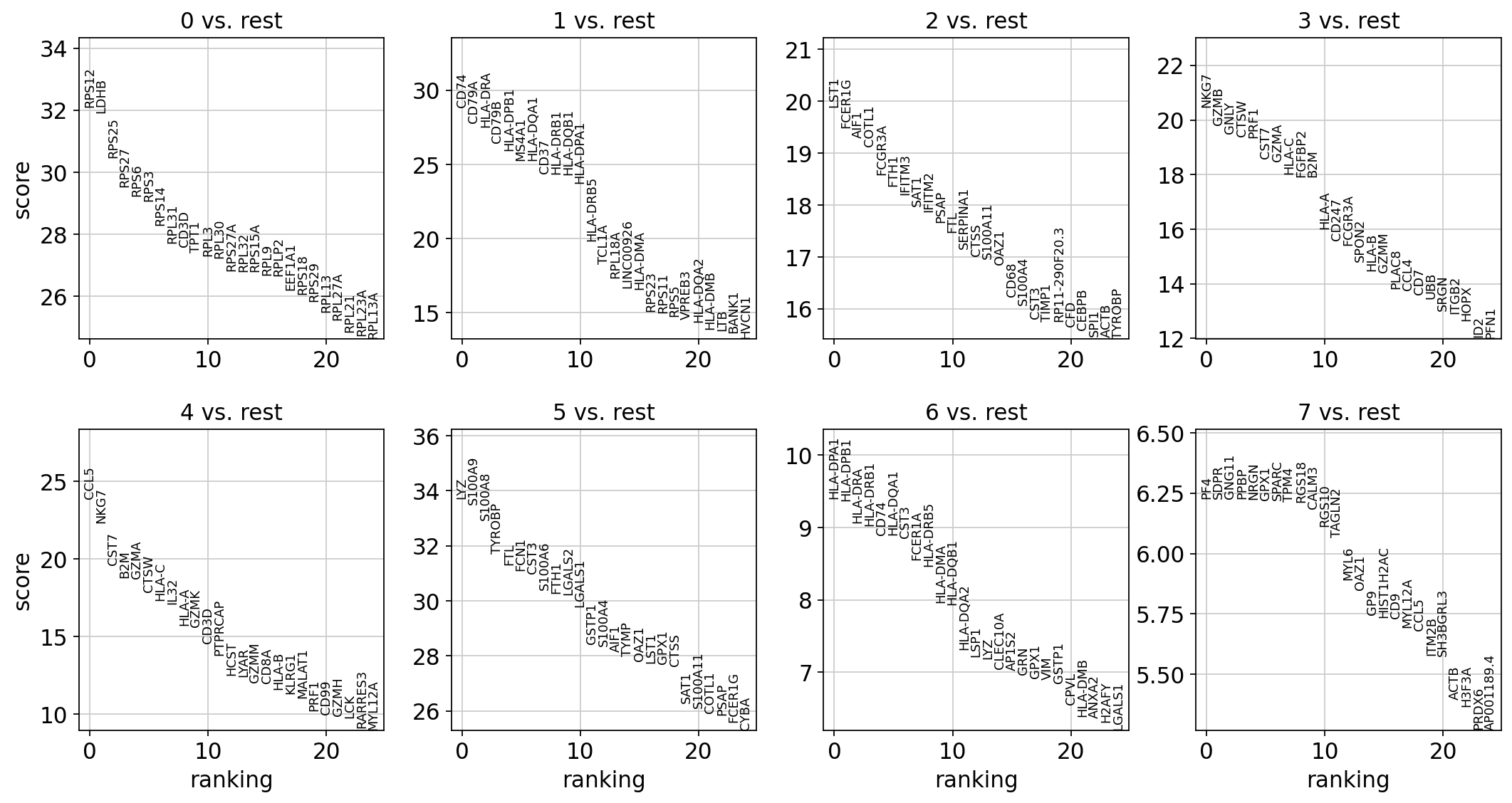
# adata.write("data/write/pbmc3k.h5ad")
# adata = sc.read_h5ad("data/write/pbmc3k.h5ad")
adata
# adata.obs["sample"] = "pbmc3k"
# adata.obs['leiden'].unique()
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden', 'sample'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'hvg', 'leiden', 'leiden_colors', 'leiden_sizes', 'log1p', 'neighbors', 'paga', 'pca', 'umap', 'tsne', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
obsp: 'connectivities', 'distances'
输出marker genes的统计检验结果
可以按照分组情况查看各个cluster的marker genes的差异性。
sc.get.rank_genes_groups_df(adata, group = '1')
| names | scores | logfoldchanges | pvals | pvals_adj | |
|---|---|---|---|---|---|
| 0 | CD74 | 28.874624 | 4.079535 | 2.487145e-183 | 3.410871e-179 |
| 1 | CD79A | 27.834063 | 7.749746 | 1.679730e-170 | 1.151791e-166 |
| 2 | HLA-DRA | 27.533710 | 4.887034 | 6.935111e-167 | 3.170270e-163 |
| 3 | CD79B | 26.463242 | 5.518004 | 2.569135e-154 | 8.808280e-151 |
| 4 | HLA-DPB1 | 25.923950 | 4.065814 | 3.577195e-148 | 9.811532e-145 |
| ... | ... | ... | ... | ... | ... |
| 13709 | IL32 | -16.639757 | -4.344168 | 3.590714e-62 | 2.462153e-59 |
| 13710 | ANXA1 | -16.926432 | -3.686769 | 2.872739e-64 | 2.188708e-61 |
| 13711 | S100A6 | -19.280443 | -3.159241 | 7.840220e-83 | 7.168052e-80 |
| 13712 | TMSB4X | -20.735228 | -0.862879 | 1.666637e-95 | 1.758174e-92 |
| 13713 | S100A4 | -22.245899 | -3.911268 | 1.235875e-109 | 1.412399e-106 |
13714 rows × 5 columns
查看排序后的基因并转换为padas对象
列出各个cluster特征性表达基因方便后续注释。
pd.DataFrame(adata.uns['rank_genes_groups']['names']).head()
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| 0 | RPS12 | CD74 | LST1 | NKG7 | CCL5 | LYZ | HLA-DPA1 | PF4 |
| 1 | LDHB | CD79A | FCER1G | GZMB | NKG7 | S100A9 | HLA-DPB1 | SDPR |
| 2 | RPS25 | HLA-DRA | AIF1 | GNLY | CST7 | S100A8 | HLA-DRA | GNG11 |
| 3 | RPS27 | CD79B | COTL1 | CTSW | B2M | TYROBP | HLA-DRB1 | PPBP |
| 4 | RPS6 | HLA-DPB1 | FCGR3A | PRF1 | GZMA | FTL | CD74 | NRGN |
sc.pl.rank_genes_groups_violin(adata, groups = '0',n_genes = 8)
/home/woodpecker/anaconda3/envs/py3.10/lib/python3.10/site-packages/scanpy/plotting/_tools/__init__.py:1303: UserWarning: set_ticklabels() should only be used with a fixed number of ticks, i.e. after set_ticks() or using a FixedLocator.
_ax.set_xticklabels(new_gene_names, rotation="vertical")
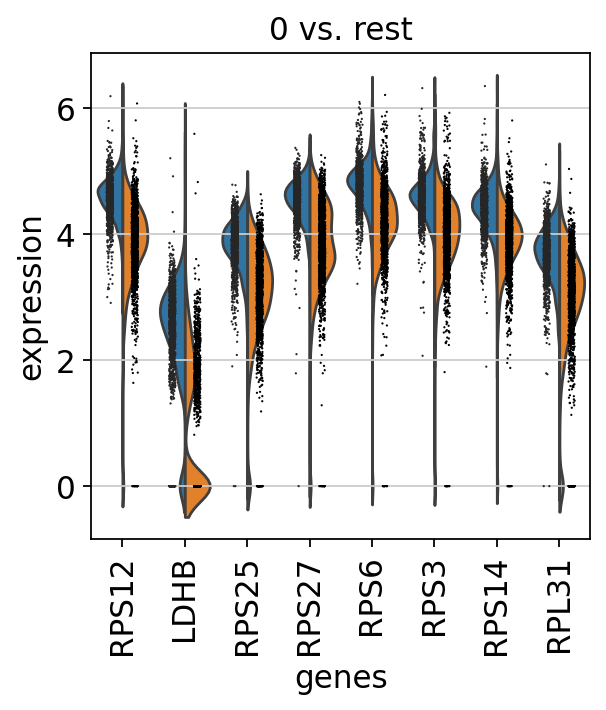
查找单个cluster与单个cluster之间差异基因的表达
则加入 reference = ‘cluster_name’参数,比如接下来比较cluster0与cluster1的差异基因表达:
sc.tl.rank_genes_groups(adata, "leiden", groups=["0"], reference="1", method="wilcoxon")
sc.pl.rank_genes_groups_violin(adata, groups = '0',n_genes=8)
ranking genes
finished: added to `.uns['rank_genes_groups']`
'names', sorted np.recarray to be indexed by group ids
'scores', sorted np.recarray to be indexed by group ids
'logfoldchanges', sorted np.recarray to be indexed by group ids
'pvals', sorted np.recarray to be indexed by group ids
'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:03)
/home/woodpecker/anaconda3/envs/py3.10/lib/python3.10/site-packages/scanpy/plotting/_tools/__init__.py:1303: UserWarning: set_ticklabels() should only be used with a fixed number of ticks, i.e. after set_ticks() or using a FixedLocator.
_ax.set_xticklabels(new_gene_names, rotation="vertical")
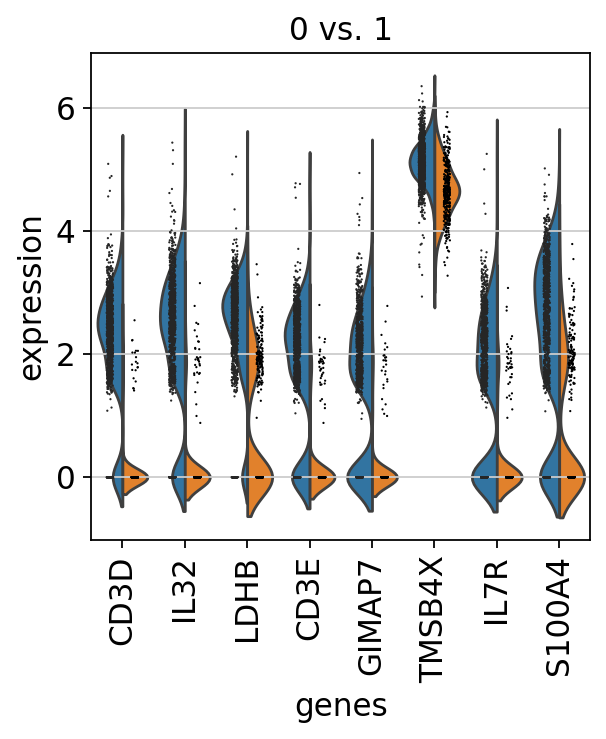
可视化单个基因在不同cluster中的表达情况
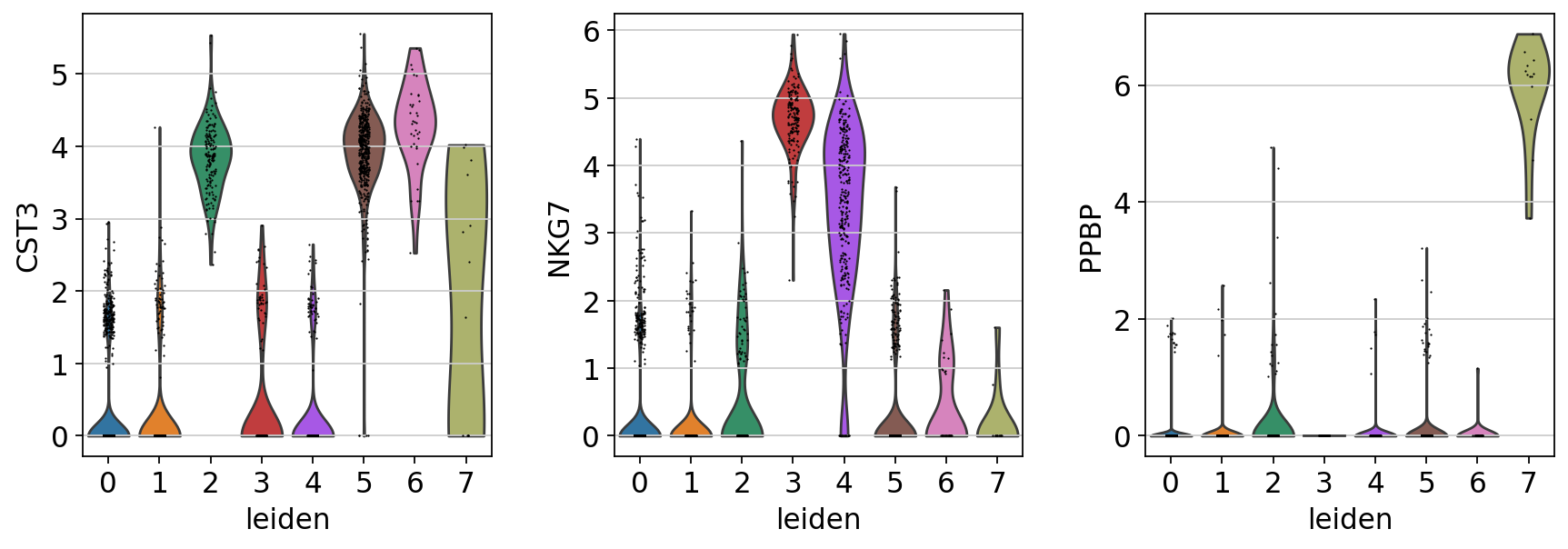
sc.pl.violin(adata, ["CST3", "NKG7", "PPBP"], groupby="leiden")
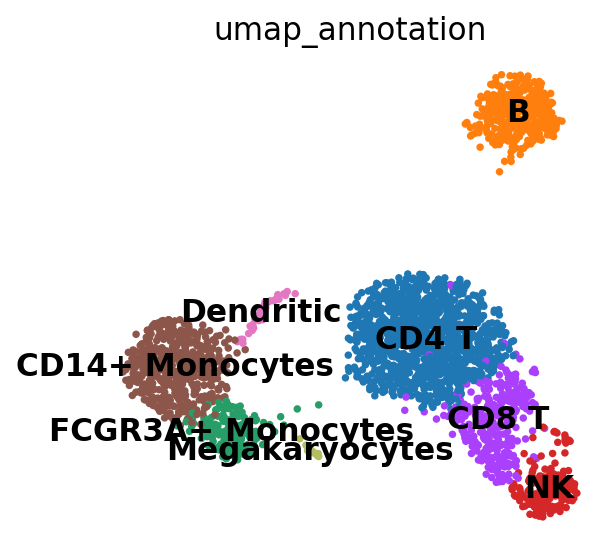
对细胞类型进行注释
new_cluster_names = [
"CD4 T",
"B",
"FCGR3A+ Monocytes",
"NK",
"CD8 T",
"CD14+ Monocytes",
"Dendritic",
"Megakaryocytes",
]
adata.rename_categories("leiden", new_cluster_names)
sc.pl.umap(
adata, color="leiden", legend_loc="on data", title="umap_annotation", frameon=False, save=".pdf"
)
WARNING: saving figure to file figures/umap.pdf
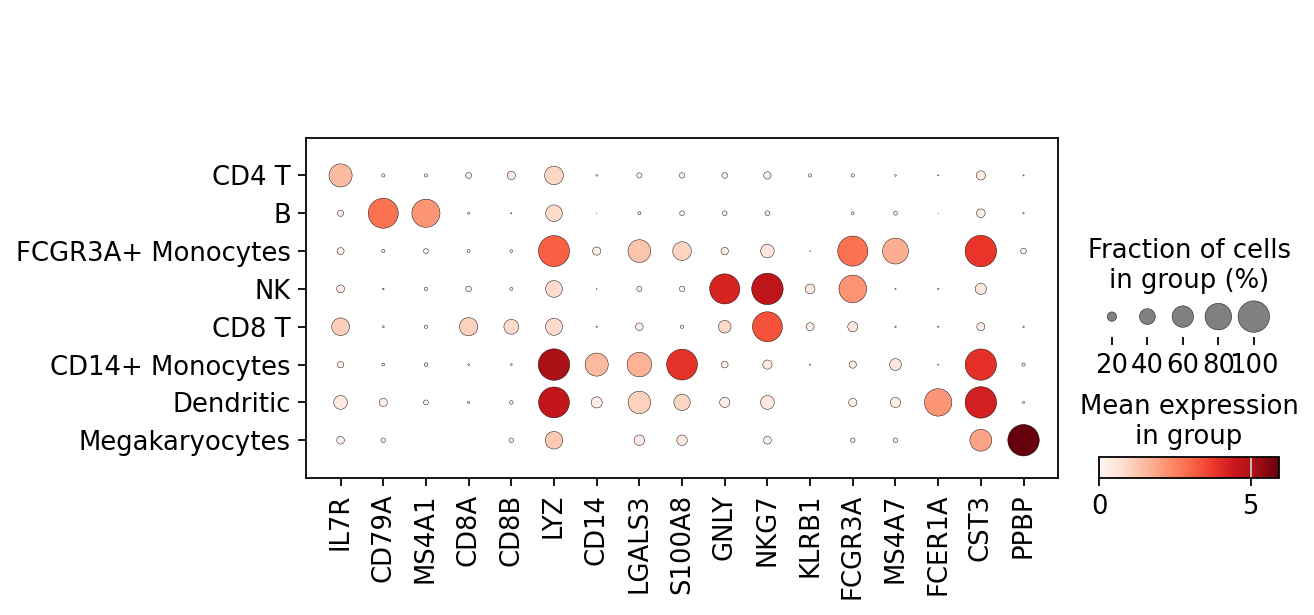
对标记基因进行可视化
marker_genes = [
*["IL7R", "CD79A", "MS4A1", "CD8A", "CD8B", "LYZ", "CD14"],
*["LGALS3", "S100A8", "GNLY", "NKG7", "KLRB1"],
*["FCGR3A", "MS4A7", "FCER1A", "CST3", "PPBP"],
]
sc.pl.dotplot(adata, marker_genes, groupby="leiden")
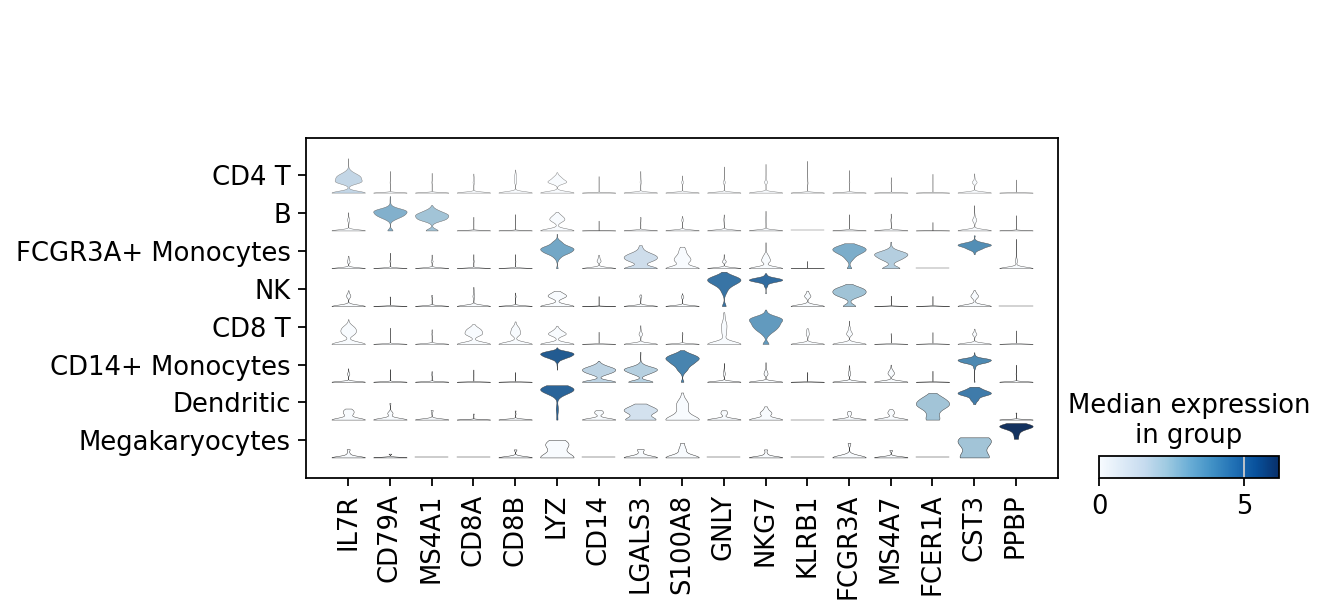
sc.pl.stacked_violin(adata, marker_genes, groupby="leiden")
概览adata目前经过计算后累积的注释信息并进行数据的保存
直接输入adata查看概览 用write保存数据,默认采用’gzip’的压缩方式,保存为.h5ad格式
adata
AnnData object with n_obs × n_vars = 2638 × 1838
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden', 'sample'
var: 'gene_ids', 'n_cells', 'mt', 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std'
uns: 'hvg', 'leiden', 'leiden_colors', 'leiden_sizes', 'log1p', 'neighbors', 'paga', 'pca', 'umap', 'tsne', 'rank_genes_groups'
obsm: 'X_pca', 'X_umap', 'X_tsne'
varm: 'PCs'
obsp: 'connectivities', 'distances'
adata.write("data/write/pbmc3k.h5ad")