Anndata数据结构学习
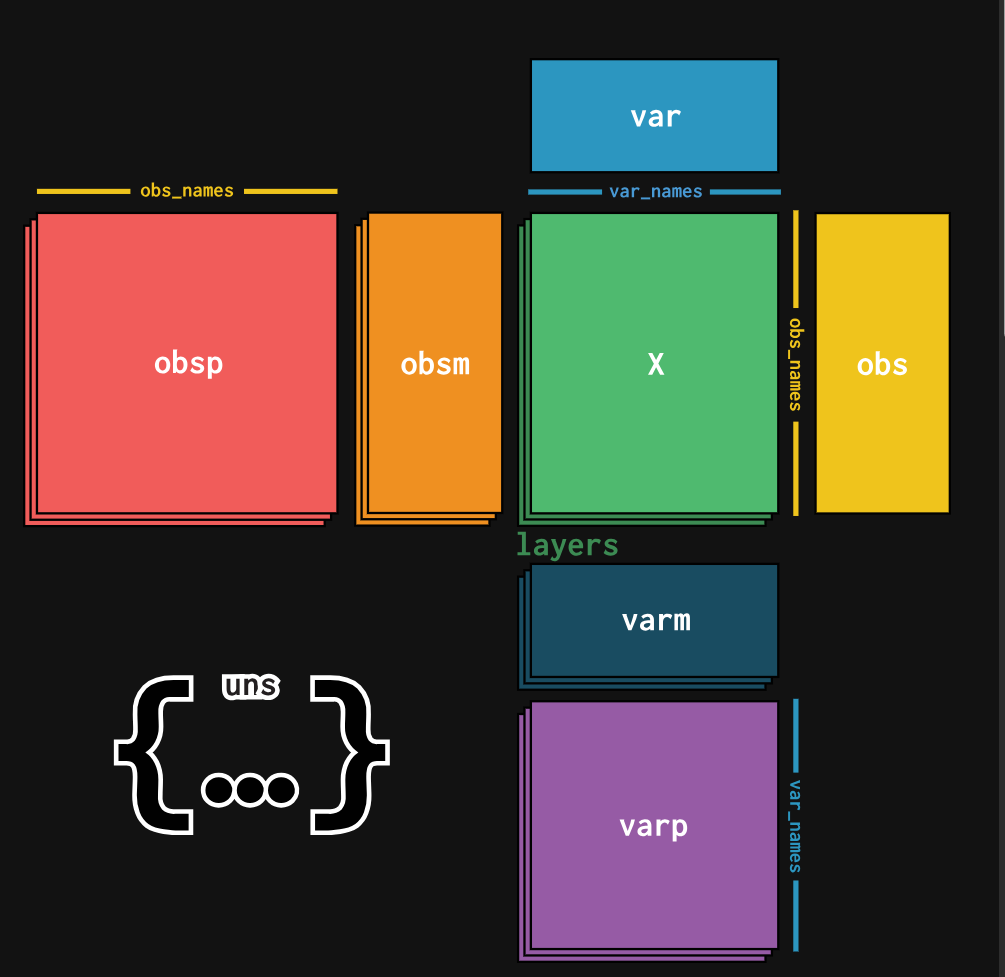
数据结构快速解读:
- X:数据矩阵,即原始数据,shape为(n_obs, n_vars)
- obs:观测数据,即样本信息,shape为(n_obs, n_obs_meta)
- var:变量数据,即特征信息,shape为(n_vars, n_var_meta)
- uns:未分组数据,即其他信息,shape为(n_uns, n_uns_meta)
- obsm:观测相关矩阵,即样本与观测相关的矩阵,shape为(n_obs, n_obs_meta)
- varm:变量相关矩阵,即特征与变量相关的矩阵,shape为(n_vars, n_var_meta)
- layers:数据层,即不同数据类型,shape为(n_obs, n_layers)
- obsp:观测相关图,即样本间的关系,shape为(n_obs, n_obs, n_obs_meta),通常是一个二维稀疏矩阵,行数和列数都与 adata.n_obs 相同,表示每个观察单元与其他观察单元之间的关系
- varp:变量相关图,即特征间的关系,shape为(n_vars, n_vars, n_var_meta),通常是一个二维稀疏矩阵,行数和列数都与 adata.n_vars 相同,表示每个变量与其他变量之间的关系
加载所需的库
加载完毕后打印目前anndata版本号
import numpy as np
import pandas as pd
import anndata as ad
from scipy.sparse import csr_matrix
print(ad.__version__)
0.10.7
初始化AnnData 让我们首先构建一个带有一些稀疏计数信息的基本AnnData对象,可能表示基因表达计数。
- 使用NumPy库中的random.poisson函数生成一个符合泊松分布的随机数矩阵。
- 1是泊松分布的lambda参数,表示该分布的均值和方差。
- size=(100, 2000)指定了生成的矩阵的形状,即100行2000列。
- 生成的矩阵中的每个元素都是从泊松分布中随机抽取的一个非负整数。
csr_matrix(…, dtype=np.float32):
这行代码将上一步生成的随机数矩阵转换为压缩稀疏行(Compressed Sparse Row)格式的矩阵。 csr_matrix是SciPy库中的一个函数,用于创建这种稀疏矩阵。
counts = csr_matrix(np.random.poisson(1, size=(100, 2000)), dtype=np.float32)
adata = ad.AnnData(counts)
adata
adata.X
<100x2000 sparse matrix of type '<class 'numpy.float32'>'
with 126382 stored elements in Compressed Sparse Row format>
adata.obs_names = [f"Cell_{i:d}" for i in range(adata.n_obs)]
adata.var_names = [f"Gene_{i:d}" for i in range(adata.n_vars)]
print(adata.obs_names[:10],"\n", adata.var_names[:10])
Index(['Cell_0', 'Cell_1', 'Cell_2', 'Cell_3', 'Cell_4', 'Cell_5', 'Cell_6',
'Cell_7', 'Cell_8', 'Cell_9'],
dtype='object')
Index(['Gene_0', 'Gene_1', 'Gene_2', 'Gene_3', 'Gene_4', 'Gene_5', 'Gene_6',
'Gene_7', 'Gene_8', 'Gene_9'],
dtype='object')
对anndata数据进行索引
可以通过索引值或者索引名来获取数据集中的数据,索引值是整数,索引名是字符串。
subdata_1 = adata[["Cell_1", "Cell_2"], ["Gene_1", "Gene_2"]]
subdata_2 = adata[[1,2], [1,2]]
data_type_1 = subdata_1
data_type_2 = subdata_2.X
data_value = subdata_2.X.toarray()
print(f"anndata对象输出:{data_type_1}\n"
f"anndata.X输出稀疏矩阵:{data_type_2}\n"
f"anndata.X.toarray()输出密集矩阵:{data_value}")
anndata对象输出:View of AnnData object with n_obs × n_vars = 2 × 2
anndata.X输出稀疏矩阵: (0, 0) 1.0
(0, 1) 2.0
anndata.X.toarray()输出密集矩阵:[[1. 2.]
[0. 0.]]
以上输出结果解读: 直接打印子集对象subdata_1,输出的是对该对象的描述,即2个观察值和2个变量。 打印subdata_2.X则是输出稀疏矩阵的形式,即矩阵的元素只有非零元素。具体为第一行第一列(0,0),其值为1.0,以及第一行第二列(0,1),其值为2.0. 打印subdata_2_X.toarray()则是将稀疏矩阵转换为密集矩阵,即numpy.ndarray的形式。其值为python中的数组[[1. 2.] [0. 0.]]
添加对齐的元数据
Observation/Variable level 所以我们有了对象的核心anndata.X,现在我们想在观察和变量级别添加元数据。这对于AnnData来说非常简单,adata. obs和adata.var都是Pandas DataFrames。 np.random.choice()是NumPy库中的一个函数,用于从给定的一维数组或列表中随机选择元素。它可以根据指定的概率从数组中抽取元素(中括号中的列表元素),也可以指定抽取的数量(size)。 adata.obs[“cell_type”] = pd.Categorical(ct): 这一行代码将ct数组转换为Pandas的Categorical数据类型,并将其作为cell_type列添加到adata.obs中。adata.obs是AnnData对象的一个属性,表示观测样本的注释信息。通过将ct列添加到adata.obs中,可以标记每个细胞的细胞类型。使用Categorical数据类型可以减少内存使用,并提高计算效率,特别是在后续处理中需要对细胞类型进行分组或统计时。
ct = np.random.choice(["B", "T", "Monocyte"], size=(adata.n_obs,))
adata.obs["cell_type"] = pd.Categorical(ct)
adata.obs
在 size=(adata.n_obs,) 中,(adata.n_obs,) 是一个包含单个元素的元组。这里之所以使用逗号,是因为在 Python 中,仅有一个元素的元组需要在元素后面加上逗号以区分它是一个元组而不是一个普通的括号表达式
用刚才添加的celltype对AnnData数据取子集
bdata = adata[adata.obs.cell_type == "B"]
bdata.obs[:10]
| cell_type | |
|---|---|
| Cell_1 | B |
| Cell_2 | B |
| Cell_9 | B |
| Cell_14 | B |
| Cell_17 | B |
| Cell_21 | B |
| Cell_23 | B |
| Cell_25 | B |
| Cell_26 | B |
| Cell_28 | B |
</div>
Observation/variable-level matrices多维元数据的嵌入
我们可能在任一级别都有多维数的元数据,例如数据的UMAP嵌入。对于这种类型的元数据,AnnData具有. obsm/.varm属性。我们使用键来标识我们插入的不同矩阵。.obsm/.varm的限制是.obsm矩阵的长度必须等于观察值的数量n_obs, .varm矩阵的长度必须等于变量个数n_vars
adata.obsm["X_umap"] = np.random.normal(0, 1, size=(adata.n_obs, 2))
adata.varm["gene_stuff"] = np.random.normal(0, 1, size=(adata.n_vars, 5))
print(f"{adata.obsm}\n{adata.varm}")
AxisArrays with keys: X_umap
AxisArrays with keys: gene_stuff
如果针对于obs或var的元数据是多维的,则储存在.obsm和.varm中,键名为字符串。
非结构化元数据
AnnData有. uns,它允许任何非结构化元数据。这可以是任何东西,比如列表或字典,其中包含一些对我们的数据分析有用的一般信息
adata.uns["random"] = [1, 2, 3]
adata.uns["nickname"] = ["haha", "hehe", "hoho"]
adata.uns["introduction"] = "This is a test of uns."
nickname = adata.uns["nickname"]
intro = adata.uns["introduction"]
print(f"{nickname}\n{intro}")
['haha', 'hehe', 'hoho']
This is a test of uns.
Layers
我们可能有不同形式的原始核心数据,可能一种是规范化的,一种不是。这些可以存储在AnnData的不同层中。例如,让我们对原始数据进行日志转换并将其存储在一个层中
adata.layers["log_transformed"] = np.log1p(adata.X)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random', 'nickname', 'introduction'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
np.log1p(adata.X): 这里使用了 numpy 库中的 log1p 函数。log1p 函数计算的是输入数组中每个元素加 1 后的自然对数。在单细胞数据处理中,对表达数据进行对数转换是一个常见的预处理步骤。这是因为单细胞表达数据通常具有高度的偏斜分布,即大多数细胞的表达量很低,而少数细胞的表达量非常高。对数转换可以使得数据分布更加接近正态分布,有助于后续的数据分析和建模。
转换为DataFrames
我们还可以要求AnnData从其中一个层返回给我们一个DataFrame: obs_names/。var_names被用于创建这个Pandas对象。
adata.to_df(layer="log_transformed").head()
| Gene_0 | Gene_1 | Gene_2 | Gene_3 | Gene_4 | Gene_5 | Gene_6 | Gene_7 | Gene_8 | Gene_9 | ... | Gene_1990 | Gene_1991 | Gene_1992 | Gene_1993 | Gene_1994 | Gene_1995 | Gene_1996 | Gene_1997 | Gene_1998 | Gene_1999 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell_0 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.000000 | 0.693147 | ... | 0.000000 | 0.000000 | 0.000000 | 0.693147 | 0.693147 | 0.693147 | 1.098612 | 1.386294 | 1.098612 | 1.098612 |
| Cell_1 | 1.386294 | 0.693147 | 1.098612 | 0.000000 | 0.693147 | 0.000000 | 1.609438 | 0.693147 | 1.098612 | 1.098612 | ... | 0.000000 | 0.693147 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 0.693147 | 1.386294 |
| Cell_2 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 0.000000 | 1.098612 | 0.693147 | 0.000000 | 0.000000 | ... | 0.693147 | 1.098612 | 1.098612 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 0.693147 | 0.693147 | 1.098612 |
| Cell_3 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.098612 | 1.098612 | 0.693147 | 0.000000 | 1.386294 | 1.098612 | ... | 1.098612 | 0.693147 | 1.386294 | 1.098612 | 0.693147 | 1.098612 | 0.693147 | 1.098612 | 1.386294 | 0.693147 |
| Cell_4 | 0.693147 | 0.000000 | 0.693147 | 0.693147 | 1.098612 | 0.000000 | 1.098612 | 0.693147 | 0.693147 | 1.386294 | ... | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 1.609438 | 0.693147 | 0.693147 | 0.000000 |
5 rows × 2000 columns
</div>
将结果写入磁盘
AnnData带有自己的基于HDF5的持久文件格式:h5ad。如果具有少量类别的字符串列尚未分类,AnnData将自动转换为分类。
adata.write('my_results.h5ad', compression="gzip")
!h5ls 'my_results.h5ad'
X Group
layers Group
obs Group
obsm Group
obsp Group
uns Group
var Group
varm Group
varp Group
关于anndata数据的索引,还可以混合索引值和索引名,例如:
subdata = adata[["Cell_4", "Cell_2"], :3]
subdata
View of AnnData object with n_obs × n_vars = 2 × 3
obs: 'cell_type'
uns: 'random', 'nickname', 'introduction'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'