fgsea快速富集分析
官方教程用的是示例数据 examplePathways 和 exampleRanks,虽然照着他的代码跑下来了,但是将数据应用到后面就会报错,相信也有和我一样的,这里其实是有个坑。所以这次用自己的数据跑一遍,避免跳坑。
首先是加载包,fgsea包是建立在data.table数据基础上的,然后为了更方便读取gmt文件,还需要qusage包,包的安装不再赘述。
rm(list = ls());gc()
library(qusage)
library(fgsea)
library(data.table)
数据读取
数据读取分两部分,gmt文件用 qusage包的read.gmt()函数,一行代码搞定,返回的正是fgsea需要的list格式的数据,这和clusterProfiler包中返回一个data.frame是不同的。
geneset <- read.gmt("c2.cp.wikipathways.v2023.2.Hs.symbols.gmt")
class(geneset) # 查看返回数据类型
lapply(geneset[1:3],head) #查看前3个list
读取差异基因,选取log2FoldChange列并以基因名命名,按照降序排列。
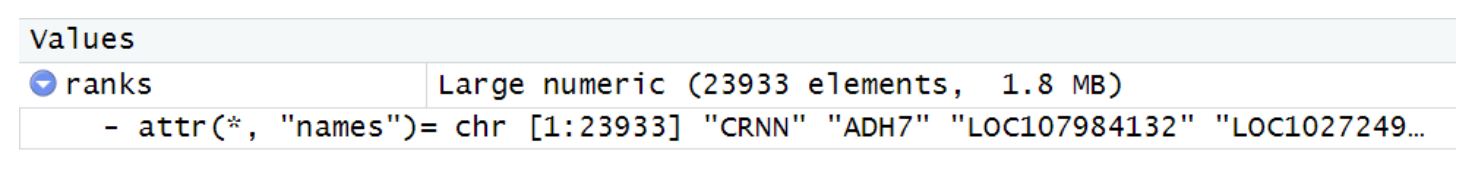
gene <- fread('different_gene.csv')
ranks <- gene[,log2FoldChange]
names(ranks) <- gene$V1
ranks <- ranks[order(ranks, decreasing = TRUE)]
这里要注意,最后输入fgsea主要函数中进行分析的的ranks必须是一个数值型向量,并且需要用gene名对每个元素进行命名,数据类型不能是data.farame或data.table,也不是list。这是我踩坑的地方。
fgsea分析
只有一行代码,有些参数基本都是默认的直接省略,有需要再去查询文档修改。富集分析完后先将富集结果进行保存,在excel中查看。
fgseaRes <- fgsea(geneset, ranks, minSize=15, maxSize=500)
fgseaRes <- fgseaRes[order(fgseaRes$NES, decreasing = TRUE)]
fwrite(fgseaRes, 'result_fgsea.csv')
警告的意思似乎是我们的rank中,有些基因(占0.96%)的排名出现并列,这时候软件采取的是随机的排列,警告这些可能会影响后续结果。(以上仅是个人查询网页结果,如有错误请告知)
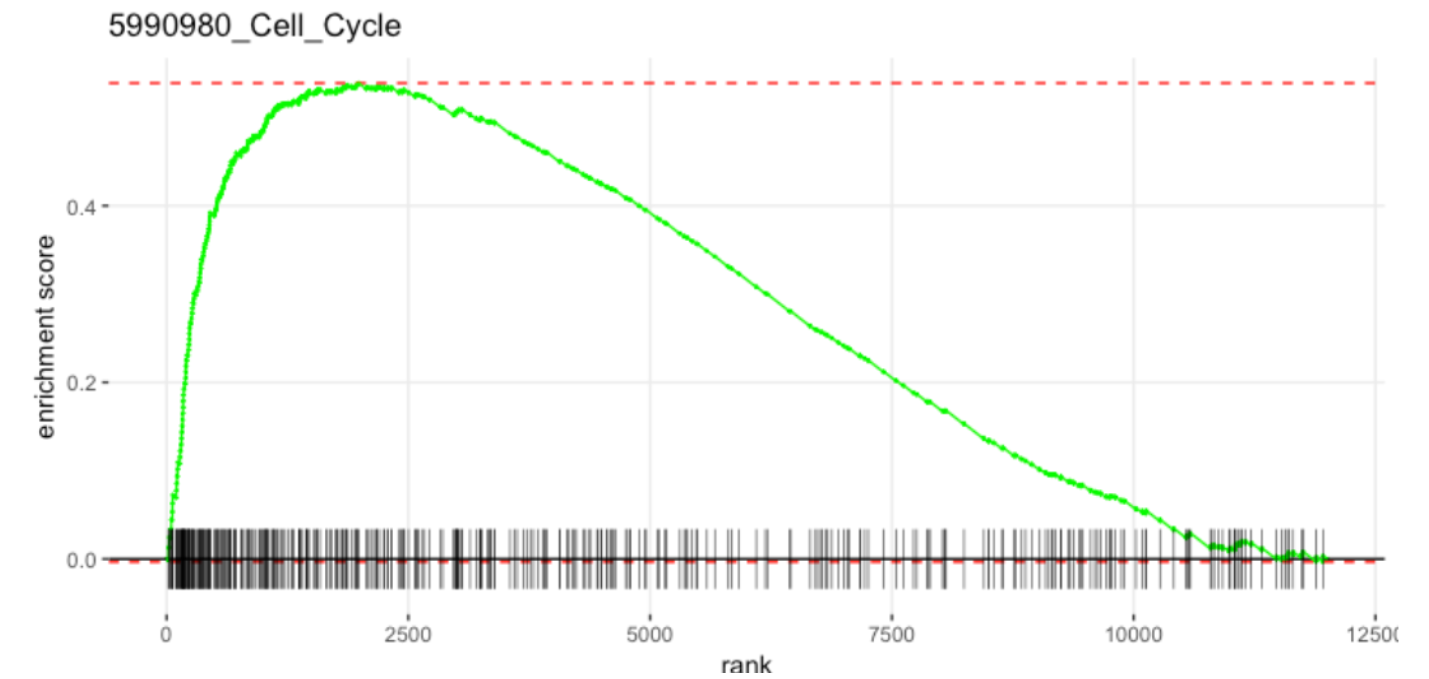
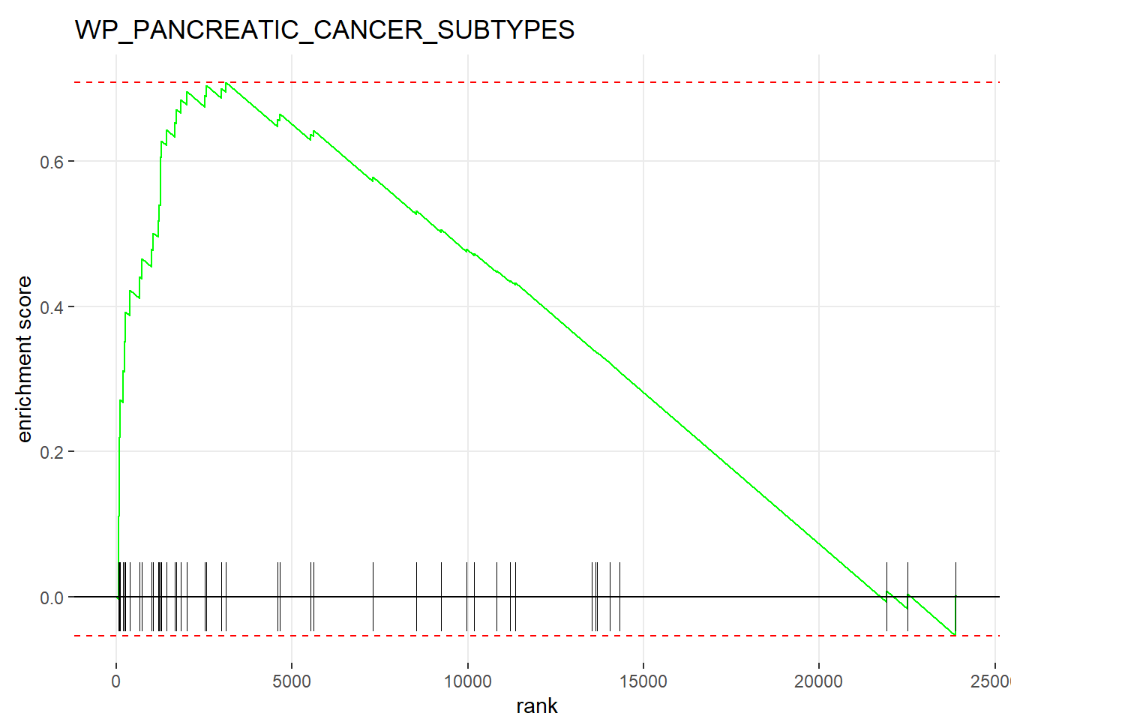
单个通路富集结果的可视化
# 绘制富集结果
library(ggplot2)
plotEnrichment(geneset[["WP_PANCREATIC_CANCER_SUBTYPES"]], ranks # 绘制富集曲线图
) + labs(title="WP_PANCREATIC_CANCER_SUBTYPES")
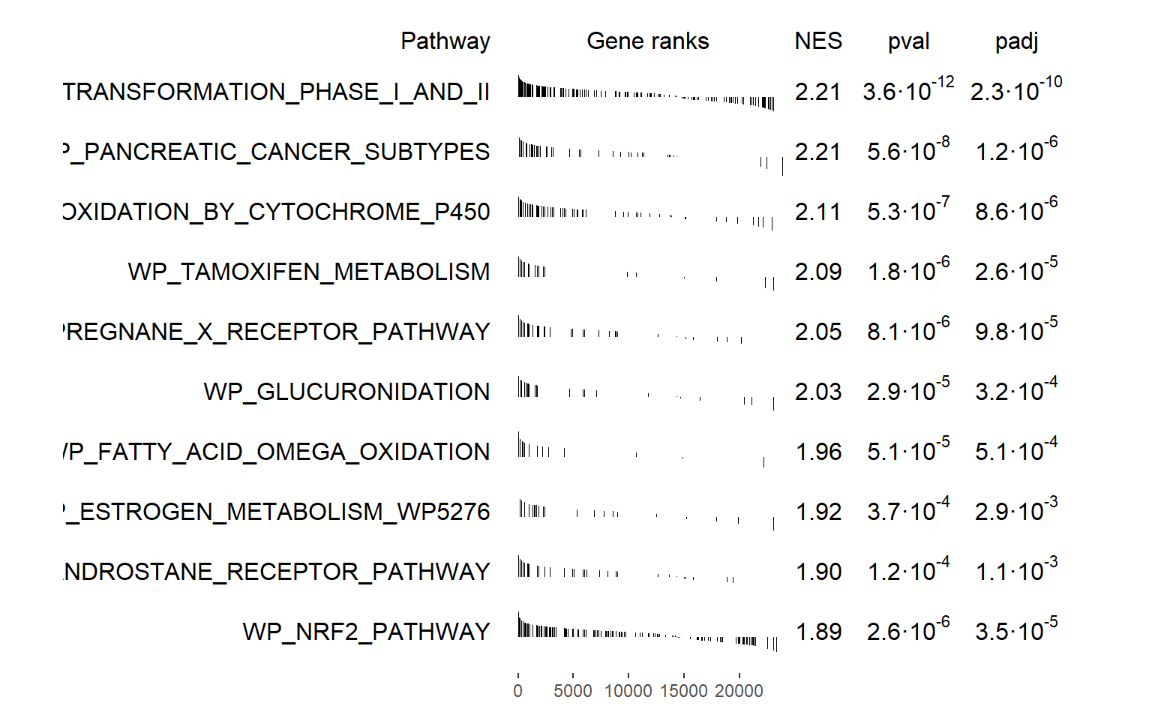
多个通路富集结果的可视化
topPathways <- fgseaRes$pathway[1:10]
plotGseaTable(geneset[topPathways], ranks, fgseaRes, gseaParam=0.5) # 这个参数通常用来控制富集分析的敏感度。